Allgemeine Information:
Bevor wir „Chiari Malformation” erklären, möchten wir den Bereich des Körpers erläutern, in dem sich eine Chiari Malformation darstellt: Bei einer normalen Entwicklung des Schädels und des Hirns ist alles an seinem Platz und erfüllt dort seine Funktion. Groß- und Kleinhirn sind so angeordnet, das sie im Schädel eine feste Stelle haben und durch die Schädelknochen nicht beeinträchtigt oder beschränkt sind.
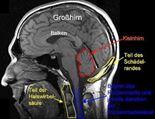
Abbildung: Normaler Schädel mit normalen Organanordnungen
Das Rückenmark hat seinen Ursprung im Gehirn und erstreckt sich im Rückenmarkskanal bis hin zum ersten/zweiten Lendenwirbel. So stellt die Wirbelsäule eine schützende Hülle für das Rückenmark dar.
Nervenwasser (Liquor) wird in Gehirnkammern (Ventrikel) ständig gebil- det, durchfließt anschließend das Kammersystem und umspült das Gehirn.
Am Ende des Kleinhirns wird es durch die hintere Schädelgrube rhythmisch- pulsierend in den Rückenmarkskanal abgegeben und umspült dort das Rückenmark.
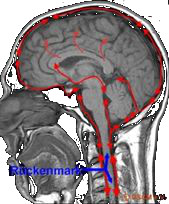
Abbildung: Zirkulation des Liquors im Schädel und Rückenmarkskanal.
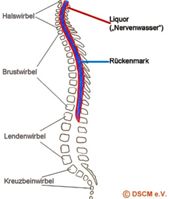
Abbildung: Schematische Darstellung Wirbelsäule mit Rückenmark und Liquor.
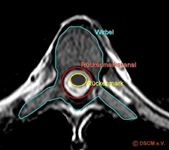
Abbildung: Horizontaler Querschnitt durch einen Brustwirbel mit Einzeichnung von Rückenmark und –kanal.
In der Mitte des Rückenmarks ist ein Zentralkanal, der von der grauen Substanz, also einem Strang von Nervengewebe, umgeben ist. Die graue Substanz ist eingebettet in weißer Substanz und ummantelt von verschiedenen Häuten (weiche Hirnhaut, Spinnengewebshaut und harte Hirnhaut). Dem Rückenmark entspringen verschiedene Nerven (Spinalnerven), die motorische Signale vom Gehirn über periphere Nerven an die Haut, Knochen, Muskeln und Gelenke leiten (so, wie sie umgekehrt sensorische Informationen zum Gehirn leiten). Jeder Nerv versorgt einen begrenzten Hautbereich und eine bestimmte Muskelgruppe; Brustnerven versorgen größtenteils Haut und Muskeln des Rumpfes, wohingegen die vorderen Äste der Hals-, Lenden- und Kreuzbeinnerven komplexe Geflechte bilden, die man Plexus nennt.
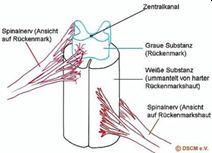
Abbildung: Vom Rückenmark ausgehende Spinalnerven (je nach Höhe des Austritts acht Halsnerven, zwölf Brustnerven, fünf Lendennerven, fünf Kreuzbeinnerven und ein Steinnerv.
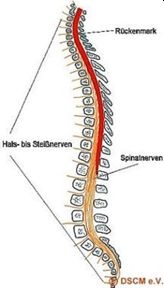
Abbildung: Schematische Darstellung des Rückenmarks mit beispielhafter, vereinfachter Zeichnung der Spinalnerven. Spinalnerven treten immer paarweise aus dem Rückenmark aus (es müssten hier also vier Spinalnerven eingezeichnet sein).
Was ist Chiari Malformation?
Die Chiari Malformation (früher auch Arnold-Chiari-Malformation oder kurz ACM genannt) gehört zu den häufigsten embryonalen Entwicklungsstörungen, 8 pro 100 000, und bildet sich in der 6.-10. Schwangerschaftswoche aus.
Bei einer Chiari Malformation kommt es zu einer knöchernen Fehlbildung des Schädelrandes und der ersten Halswirbel. Durch diese knöchernen Veränderungen findet sich unter der Schädeldecke nicht genügend Platz für einige der hinteren Hirnanteile (Kleinhirn, Kleinhirntonsillen). Aufgrund dieses Platzmangels suchen sich diese einen neuen Platz und dieser ist dann der Übergang zwischen Schädel und Wirbelsäule. Durch das sogenannte Hinterhauptsloch ragen die verdrängten Hirnanteile in den Rückenmarkskanal oder treten nach Außen. Aufgrund der unterschiedlichen Ausprägungen wird Chiari Malformation in die Typen I, II, III, IV unterteilt.
Beim Typ I liegt eine Verlagerung der Kleinhirntonsillen vor, zumeist mit einer Syringomyelie (also einer sich stets verändernden Hohlraumbildung im Rückenmark, siehe dort) einhergehend.
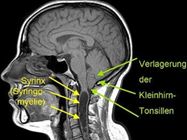
Abbildung: Chiari Malformation Typ I (gekennzeichnet durch grüne Pfeile), hier zusätzlich mit einer Syrinx (Hohlraum im Rückenmark), also einer Syringomyelie.
Gelegentlich können auch knöcherne Fehlbildungen von Schädelknochen und Wirbeln bei der Chiari Malfomation Typ I diagnostiziert werden. Diese Form der Chiari Malformation ist meist bis zum zweiten oder dritten Lebensjahr voll ausgebildet, obgleich die gesundheitlichen Beschwerden oft erst um das dreißigste Lebensjahr deutlich feststellbar sind.
Beim Typ II liegt eine leichte bis starke Verlagerung des Kleinhirns kombiniert mit einer Fehlbildung des Hirnstammes vor.
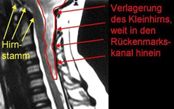
Abbildung: Verlagerung von Kleinhirntonsillen, Kleinhirnwurm und Hirn- stamm in den Rückenmarkskanal mit Erweiterung des großen Hinterhaupt- lochs (nicht eingezeichnet).
Hinzu kommt, dass im verlagerten Hirngewebe Nervenzellen absterben. Bei ca. 80 % der von Chiari Malformation Typ II betroffenen sind die Gehirnkammern (Ventrikel), die das Hirn- und Nervenwasser bilden, krankhaft verändert, so dass es zu einem Stau des Hirnwassers kommt, durch den der Schädel im Volumen stärker wächst – man bezeichnet dies als Hydrocephalus (umgangssprachlich auch Wasserkopf genannt).
Selten ist der Typ III anzutreffen, der weitgehend mit Typ II identisch ist, bis auf die komplette Verlagerung des Kleinhirns und/oder des Hirnstammes in den Nackenbereich. Diese Gewebeverlagerung kann bei einer Lücke in den Schädelknochen austreten, was als Enzephalozele bezeichnet wird.
Noch seltener ist der Typ IV. Er beschreibt eine genetisch bedingte Unterentwicklung (Hypoplasie) des Kleinhirns bei kleinerer hinterer Schädelgrube, die im Wesentlichen mit Hirnwasser gefüllt ist.
Ursachen:
In der Regel ist eine Chiari Malformation angeboren und folglich eine Fehlbildung in den Wachstumsphasen des Fötus. Die Chiari Malformation wird nicht vererbt.
In seltenen Fällen ist eine Chiari Malformation erworben, zum Beispiel wenn ein Hirntumor oder eine schwere Geburtsverletzung das Kleinhirn in den Rückenmarkskanal drückt.
Symptome und Verlauf:
Die Symptome sind abhängig vom Typ der Chiari Malformation und weiterhin, ob zusätzliche Erkrankungen wie Syringomyelie oder Spina Bifida (offener Rücken).
Ansonsten kann man das Beschwerdebild wie folgt skizzieren:
• Kopf- und Nackenschmerzen
• Kompressionsyndrom
• Schlaffe Lähmungen, inkomplette Lähmungen der Arme und Beine
• Empfindungsstörungen, sensorische Defizite, Koordinationsstörungen
• Augenrollen und -verdrehen (Nystagmus)
• Sprechstörungen
• Verminderter Schluckreflex mit Schluckbeschwerden
• Atembeschwerden bis hin zum Verschlucken von Flüssigkeiten
• Pfeifende Atemgeräusche bei Verengung oder Verlagerung der oberen Luftwege
• Überstreckung des Rumpfes und der Arme und Beine
Diagnose:
Die Diagnose einer Chiari Malformation erfolgt mit Hilfe der Computertomographie oder zur detaillierteren Darstellung mit Hilfe der Magnetresonanztomographie (MRT, auch Kernspintomographie genannt), einem bildgebenden Verfahren, bei dem keine Röntgenstrahlung verwendet wird. Bei den Aufnahmen des Schädels, des Gehirns und des Rückenmarkkanals sind sowohl die für die Chiari Malformation charakteristischen Veränderungen im Schädel- als auch im Hirnbereich eindeutig darstellbar.
Auch Nebenbefunde, wie das Bestehen einer Syrinx ist in Lage und Ausdehnung feststellbar. Durch Anwendung von Kontrastmittel, das während der Untersuchung injiziert wird, können sonstige krankhafte Veränderungen wie Tumore sichtbar gemacht werden.
Um die Zirkulation des Nervenwassers im Schädel bzw. im Rückenmarkskanal darzustellen, ist man bei spezialisierten Kliniken in der Lage, mit einer speziellen MRT-Untersuchung den Fluß aufzuzeichnen. Dabei wird die Pulsation des Nervenwassers in Relation zum Herzschlag des Untersuchten angezeigt. Erfahrene und exakte Analyse der so gewonnenen Darstellung des Nervenwasserflusses lassen selbst kleinste Verklebungen mit Zirkulationsbeeinträchtigung des Nervenwassers erkennen.
Eine Entnahme von Nervenwasser aus dem Rückenmarkskanal (Lumbalpunktion) kann wichtige Hinweise auf andere krankhafte Veränderung im zentralen Nerven- system geben, sollte aber nur nach vorheriger Untersuchung des Kopfes mittels Computer- oder Magnetresonanztomografie erfolgen, um vor Entnahme von Nervenwasser klare Befunde über die intrakraniellen Druckverhältnisse zu haben.
Bei Föten stehen pränataldiagnostische Sonografie-Verfahren zur Verfügung, um Chiari Malformation zu diagnostizieren.
Therapie:
Ziel einer jeden Behandlung der Chiari Malformation ist, die Fehlbildung und deren Auswirkungen zu beseitigen.
Beim Typ I geschieht dies durch eine Operation, bei der das große Hinterhauptloch erweitert und etwaige knöcherne Fehlbildungen des Schädels korrigiert werden. Oft bildet sich hierdurch auch eine etwaige Syringomyelie zurück.
Beim Typ II wird zunächst operativ eine Abflußmöglichkeit für das sich stauende Hirnwasser geschaffen und ansonsten verfahren wie beim Typ I.
Bei den Typen III und IV sind Lebensfähigkeit und Operabilität sehr stark von den Umständen des Einzelfalles abhängig, sodass hier keine Aussagen über eine Behandlung getroffen werden können.
In jedem Fall aber sollten Diagnose und Behandlung von sehr erfahrenen Neurochirurgen vorgenommen werden.
Mila & Paul (Deutsch)
Die Erkrankung